
Dissociation of cyclopropane in double ionization continuum

Abstract
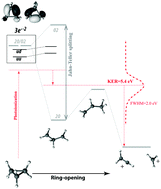
Dissociative double photoionization of cyclopropane is studied in the inner-valence region using tunable synchrotron radiation. With the aid of ab initio quantum chemical calculations the energies of dication states and their favoured fragmentation pathways are determined. These are compared to the experimental appearance energies of two-body fragmentation processes and to the kinetic energy released upon dissociation. Photon energy dependent state-selective dissociation in the 25–35 eV range is found. Calculations of dissociation pathways suggest that cyclopropane ring-deformation is selectively triggered at certain photon energies. The calculations suggest that initial ring deformation essentially determines the population of different dication states that function as gateways for particular dissociation channels. The measurements show that stepwise ionization processes populate dissociative 3e′−2 states via ring-opening and Jahn–Teller active states at photon energies below the double-ionization threshold. For energies above the double-ionization threshold the kinematics indicate that double ionization takes place predominantly within the Franck–Condon region populating 3e′−1 1e′′−1 states.
- This article is part of the themed collection: XUV/X-ray light and fast ions for ultrafast chemistry
 Please wait while we load your content...
Please wait while we load your content...

