
The role of the long-range tail of the potential in O2 + N2 collisional inelastic vibrational energy transfers
 *a
Fernando
Pirani,
b
*a
Fernando
Pirani,
b
Abstract
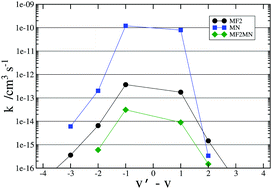
In the study of non-reactive energy transfer between O2 and N2 molecules bearing different vibrationally excited states we have faced the problem of selecting a proper formulation of the interaction. To this end we have compared the values of the related observables computed either on a potential energy surface globally fitted to very large ab initio potential energy values [Varga et al., J. Chem. Phys., 2016, 144, 024310] or on two more traditional ones formulated as a combination of an intra- and inter-molecular model component of the interaction (and based on a different combined use of experimental and ab initio information) [Garcia et al., J. Phys. Chem. A, 2016, 120, 5208] in order to enforce an appropriate modelling of the long-range tail of the potential, crucial for the description of inelastic vibrational energy transfer. A detailed graphical analysis of the potential plus a quantitative analysis of the computed opacity functions, of the state-to-state rate coefficients, of the second virial coefficient and of the integral non-reactive cross section allowed us to conclude that the model formulation of the interaction has to be preferred for non-reactive studies of the O2 + N2 energy transfer processes in thermal and subthermal regimes.
 Please wait while we load your content...
Please wait while we load your content...

