
A density functional theory study on the thermodynamic and dynamic properties of anthraquinone analogue cathode materials for rechargeable lithium ion batteries†
 a
Liu-Bin
Zhao
*a
a
Liu-Bin
Zhao
*a
Abstract
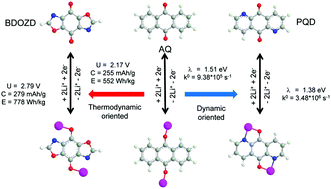
Organic redox compounds have become the emerging electrode materials for rechargeable lithium ion batteries. The high electrochemical performance provides organic electrode materials with great opportunities to be applied in electric energy storage devices. Among the different types of organic materials, conjugated carbonyl compounds are the most promising type at present, because only they can simultaneously achieve, high energy density, high cycling stability, and high power density. In this research, a series of heteroatom substituted anthraquinone (AQ) derivatives were designed theoretically so that the high theoretical capacity of AQ remained. The discharge and charge mechanism as well as the thermodynamic and dynamic properties of AQ and its derivatives were investigated using first-principles density functional theory. Using heteroatom substitution, both the thermodynamic and dynamic properties of AQ as cathode materials could be largely improved. Among these conjugated carboxyl compounds, BDOZD and BDIOZD with a simultaneously high theoretical capacity and high working potential exhibit the largest energy density of about 780 W h kg−1, which is 41% larger than that of AQ. The PQD with the smallest value of λITo gives the largest charge transfer rate constant, which is about four times as large as the prototype molecule, AQ. The most interesting finding is that the lithium ion transfer plays a very important role in influencing both the discharge potential and electrochemical charge transfer rate. The present study illustrates that theoretical calculations provide a highly effective way to discover potential materials for use with rechargeable lithium ion batteries.
 Please wait while we load your content...
Please wait while we load your content...

