
Insights into the thermoelectric properties of SnSe from ab initio calculations†

Abstract
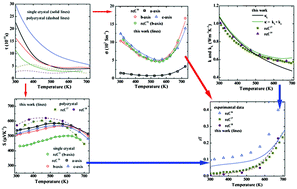
The thermoelectric properties of SnSe are studied by first-principles methods using an original methodology. We computed first the electronic structure of the system, which justifies its macroscopic anisotropy; the inclusion of van der Waals dispersive corrections improves the agreement of the structural parameters with experiments. The Seebeck coefficient and the electrical and thermal conductivities of single crystals and polycrystals are subsequently described in good agreement with experimental data. As for the electrical conductivity, values calculated with a temperature-dependent relaxation time compare well with the available measurements, especially for single crystals; in contrast, a constant relaxation time suffices to describe the results for polycrystals. Based on the iterative solution of the Boltzmann transport equation for phonons, we discuss the behavior of the thermal conductivity of the system in terms of its phonon spectrum. Finally, the figure of merit of SnSe single crystals and polycrystals is calculated and correlated with the previous discussions about electrical and thermal conductivities. From these findings, possible strategies to increase the figure of merit in practice are suggested.
 Please wait while we load your content...
Please wait while we load your content...

