
Surface reactivity and vacancy defects in single-layer borophene polymorphs†

Abstract
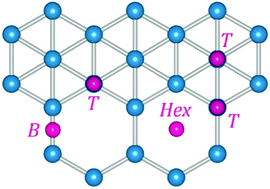
Single-layer borophene is a novel 2D material which combines high strength, light weight and metallicity. Using first-principles calculations, we systematically investigate the defect formation and surface reactivity in three major borophene polymorphs (α, β and triangular). We find that β-B is generally the most reactive borophene form, while α-B is the least reactive. In particular, there is more than 1.5 eV difference in substitutional energies for typical dopants in β-B and α-B polymorphs. Single vacancy defects can be created quite easily in all borophene sheets with formation energies (0.16 to 1.93 eV) much lower than those in graphene (7.69 eV). Adatom adsorption is exothermic and stabilizes electron-deficient boron monolayers. Many interesting properties arise from the rich structural chemistry of borophene, comprising four-, five-, and six-coordinated atoms, as well as hexagonal vacancies.
 Please wait while we load your content...
Please wait while we load your content...

