
Potassium adsorption behavior on hcp cobalt as model systems for the Fischer–Tropsch synthesis: a density functional theory study†

Abstract
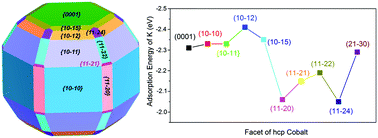
Potassium (K), an important impurity in syngas from biomass, can have a large influence on the activity and selectivity of cobalt-based Fischer–Tropsch synthesis (FTS) catalysts in Biomass to Liquids (BTL) processes. In this work, the potassium adsorption behavior on hcp cobalt was systematically studied using density functional theory. The surface energy calculations and Wulff construction of the equilibrium shape of hcp cobalt showed it is dominated by 10 facets. The interaction of K with these facets has been investigated. The results showed that the stepped facet (10−12) has the highest K adsorption energy of −2.40 eV. The facets (0001), (10−10), (10−11), (10−15), and (21−30) also showed relatively high K adsorption energies in the range of −2.28 to −2.34 eV. The corrugated facets exhibited comparatively lower K adsorption energies (−2.04 to −2.18 eV), and would be less favorable for K adsorption. It was also found that the adsorption properties depend on coverage, where the K adsorption energy decreased with increasing coverage. Diffusion energy barrier calculations indicated that K was mobile on typical facets (0001) and (10−11) with very low diffusion barriers (<0.15 eV). On stepped facets, although K could move freely along the same step (diffusion barrier <0.01 eV), diffusion from one step to another had a significantly higher barrier of 0.56 eV. This suggested that K atoms would be mobile to some extent during FTS reaction conditions, and tend to occupy the most favorable sites independent of their initial position. The results obtained in this work provide valuable information on the interaction of K with cobalt surfaces, relevant for practical cobalt catalysts and their application in BTL processes.
 Please wait while we load your content...
Please wait while we load your content...

