
Contrasting diffusion behaviors of O and F atoms on graphene and within bilayer graphene†
 a
a
Abstract
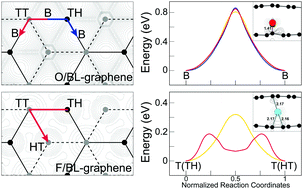
Chemical modification of graphene with adatoms is of importance for nanoelectronics applications. Based on first-principles density-functional theory calculations including van der Waals interactions, we present a comparative study of the diffusion characteristics of oxygen (O) and fluorine (F) atoms both on graphene and between the layers of bilayer graphene. We find that the calculated diffusion barrier for the O atom increases slightly from 0.81 eV on graphene to 0.85 eV within bilayer graphene, while that for the F atom largely decreases from 0.30 eV on graphene to 0.18 eV within bilayer graphene. Such contrasting behaviors of the O and F diffusions within bilayer graphene can be traced to their different bonding natures: i.e., the O adatom that shows strongly covalent C–O–C bonding on the bridge site of the C–C bond diffuses on one graphene layer with a slight interference of the other layer, while the F adatom that shows semi-ionic F–C bonding on top of a C atom easily diffuses by hopping between two graphene layers by accepting more electron charges from the two layers. The present findings have important implications for the understanding of the diffusion processes of F and O atoms on graphene and within bilayer graphene.
 Please wait while we load your content...
Please wait while we load your content...

