
A DFT study of the interplay between dopants and oxygen functional groups over the graphene basal plane – implications in energy-related applications
 *a
*a
Abstract
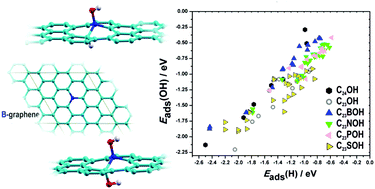
Understanding the ways graphene can be functionalized is of great importance for many contemporary technologies. Using density functional theory calculations we investigate how vacancy formation and substitutional doping by B, N, P and S affect the oxidizability and reactivity of the graphene basal plane. We find that the presence of these defects enhances the reactivity of graphene. In particular, these sites act as strong attractors for OH groups, suggesting that the oxidation of graphene could start at these sites or that these sites are the most difficult to reduce. Scaling between the OH and H adsorption energies is found on both reduced and oxidized doped graphene surfaces. Using the O2 molecule as a probe we show that a proper modelling of doped graphene materials has to take into account the presence of oxygen functional groups.
 Please wait while we load your content...
Please wait while we load your content...

