
Theoretical analysis of electrochromism under redox of bis(3-thienyl)/(2-thienyl)hexafluorocyclopentene: effects of charged and substituted systems†
 *a
*a
Abstract
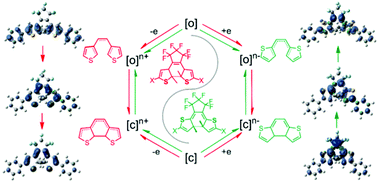
Electrochromism with the ring-closing or ring-opening isomerization of substituted and unsubstituted bis(3-thienyl)/(2-thienyl)hexafluorocyclopentene is discussed using the DFT method. In the neutral ground state, bond making and breaking between two reactive C atoms on thienyls are thermodynamically forbidden. Under redox conditions, the gain or loss of electrons can have a significant effect on the frontier molecular orbital distribution of both open- and closed-ring isomers, particularly in reactive sites. Corresponding structural changes show a trend toward isomerization. The reaction energy barrier shows greater reduction for dication than monocation and even becomes barrierless for dianion. During the isomerization in different states, the conjugated system switches distinctively, which is attributed to the special redistribution of molecular orbitals and spin population in each state. In monocation and monoanion, for the involvement of a single electron, isomerization is inclined to proceed sequentially between right and left thienyls, whereas it becomes synchronous in dication. The direction depends on the stabilization achieved by the formation of a global conjugated system and more average spin population on the molecule. The effect of substituents on thienyls is demonstrated in the promotion of the extent of conjugation and the determination of the spin population level on the reactive C atoms. Moreover, according to their electron-donating and withdrawing abilities, they can kinetically support or suppress the electron transfer pattern in the process from isomer to transition state, which leads to the control of reaction efficiency.
 Please wait while we load your content...
Please wait while we load your content...

