
Excited-state E → Z photoisomerization mechanism unveiled by ab initio nonadiabatic molecular dynamics simulation for hemithioindigo–hemistilbene†

Abstract
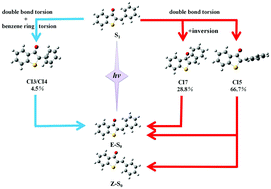
The Zhu–Nakamura formulas based on on-the-fly trajectory surface hopping dynamics simulations at the two-state-averaged CASSCF level were employed to investigate the E → Z photoisomerization mechanisms of hemithioindigo–hemistilbene (HTI) upon S1 excitation. Seven conical intersections were observed along the isomerization pathways, which were composed of double bond torsion, benzene ring torsion, inversion and pyramidalization motions, and only three of them were found to play a role in the dynamics simulations started at S1E-HTI. The dominant isomerization pathway proceeds via central double bond torsion together with pyramidal and tilt motions to some extent (hop via CI5) and accounts for all the reactive trajectories. On the other hand, the two pathways that involve the conical zones lie in the vicinity of the E-form Franck–Condon region (CI7) and proceed along the combined central double bond and benzene ring torsion route (CI3/CI4) with generation of the E products. Within the 332 simulated trajectories, 66 hop to the ground state and only 19 switch to the Z product. The estimated quantum yield of 0.057 (19 in 332) agrees well with the reported experimental value of 0.053 ± 0.016. The excited-state lifetimes span a wide region from hundreds of femtoseconds to several picoseconds, depending on the time for vibrational relaxation and number of cycles for periodical mixed mode torsion.
 Please wait while we load your content...
Please wait while we load your content...

