
Intrinsic point defects in buckled and puckered arsenene: a first-principles study
 *a
*a
Abstract
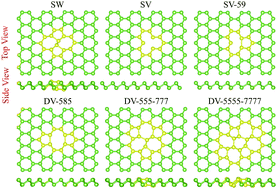
Using first-principles calculations, we study the structural, energetic, and electronic properties of various point defects in arsenene. Stone–Wales defects are found to be thermodynamically favorable and are predicted to be stable at room temperature. Defects are found to significantly influence the electronic properties in buckled phase. In particular, single vacancies generate gap states whereas strain induced states close to the valence and conduction band edges are observed for Stone–Wales and di-vacancy defects. The computed band structures of di-vacancy defects in puckered phase are less disturbed compared to the corresponding band structures in the buckled one. The influence of a hydrogen-rich atmosphere on the electronic properties of defective arsenene is also investigated. Hydrogen termination of mono/di-vacancies is an exothermic process which removes all defect induced gap states.
 Please wait while we load your content...
Please wait while we load your content...

