
A sensitive fluorescent probe for the polar solvation dynamics at protein–surfactant interfaces†
 *d
Samir Kumar
Pal
*a
*d
Samir Kumar
Pal
*a
Abstract
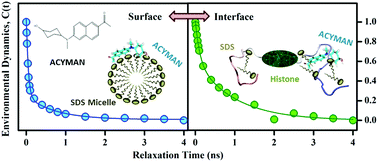
Relaxation dynamics at the surface of biologically important macromolecules is important taking into account their functionality in molecular recognition. Over the years it has been shown that the solvation dynamics of a fluorescent probe at biomolecular surfaces and interfaces account for the relaxation dynamics of polar residues and associated water molecules. However, the sensitivity of the dynamics depends largely on the localization and exposure of the probe. For noncovalent fluorescent probes, localization at the region of interest in addition to surface exposure is an added challenge compared to the covalently attached probes at the biological interfaces. Here we have used a synthesized donor–acceptor type dipolar fluorophore, 6-acetyl-(2-((4-hydroxycyclohexyl)(methyl)amino)naphthalene) (ACYMAN), for the investigation of the solvation dynamics of a model protein–surfactant interface. A significant structural rearrangement of a model histone protein (H1) upon interaction with anionic surfactant sodium dodecyl sulphate (SDS) as revealed from the circular dichroism (CD) studies is nicely corroborated in the solvation dynamics of the probe at the interface. The polarization gated fluorescence anisotropy of the probe compared to that at the SDS micellar surface clearly reveals the localization of the probe at the protein–surfactant interface. We have also compared the sensitivity of ACYMAN with other solvation probes including coumarin 500 (C500) and 4-(dicyanomethylene)-2-methyl-6-(p-dimethylamino-styryl)-4H-pyran (DCM). In comparison to ACYMAN, both C500 and DCM fail to probe the interfacial solvation dynamics of a model protein–surfactant interface. While C500 is found to be delocalized from the protein–surfactant interface, DCM becomes destabilized upon the formation of the interface (protein–surfactant complex). The timescales obtained from this novel probe have also been compared with other femtosecond resolved studies and molecular dynamics simulations.
 Please wait while we load your content...
Please wait while we load your content...

