
Modulation of the electronic and mechanical properties of phagraphene via hydrogenation and fluorination†
 *c
*c
Abstract
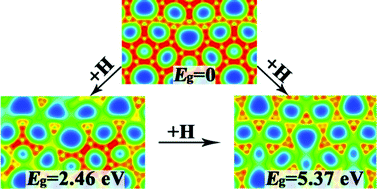
Recently, a new carbon sheet, phagraphene, was proposed by theoretical calculations [Nano Lett. 2015, 15, 6182]. In this paper, hydrogenated and fluorinated phagraphene (denoted as H-PHA and F-PHA) sheets have been systematically studied using first-principles calculations. The results of formation energy, ab initio molecular dynamics, phonon dispersion and elastic constants confirm that the modified phagraphene sheets are thermodynamically and dynamically as well as mechanically stable. We find that hydrogenation or fluorination is an effective way to modulate the bandgap, and we also find that adsorption-induced semimetal–semiconductor transition and adsorption-induced semimetal–insulator transition occur. Configuration-dependent bandgaps for partially H-PHA and configuration-independent bandgaps for fully H-PHA are determined. Adsorption-ratio-dependent bandgaps of H-PHA and F-PHA are also identified. Bandgaps calculated from HSE06 and PBE functionals of fully H-PHA are larger than those of F-PHA, and they are comparable to hydrogenated/fluorinated penta-graphene while they are larger than their corresponding graphene. Dependence of bandgaps of fully H-PHA and F-PHA on the tensile strain is investigated, and our calculations show that an insulator–semiconductor transition occurs upon increasing the tensile strain. Our results also show that the mechanical properties can be controlled using hydrogenation and fluorination. The calculations of Young's modulus and Poisson's ratio reveal that functionalized phagraphene sheets possess suitable stiffness and resistance to volume deformation, and both are smaller than those of the pristine phagraphene.
 Please wait while we load your content...
Please wait while we load your content...

