
On the validity of linear response approximations regarding the solvation dynamics of polyatomic solutes

Abstract
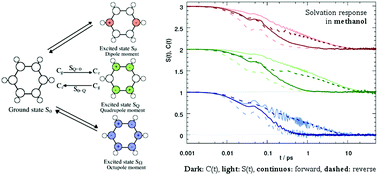
The time-dependent fluorescence of a chromophore can be calculated from either nonequilibrium simulations, or, as long as linear response theory holds true, from equilibrium solvent fluctuations in the ground or excited state if the perturbation inflicted by the chromophore is small. The assumption of Gaussian statistics, in contrast, links the nonequilibrium dynamics to solvent fluctuations solely in the excited state, as long as the energy gap distribution is Gaussian throughout the process. The validity of linear response theories on the ground and excited state surface as well as Gaussian statistics is thoroughly tested in this study by calculating the time-dependent Stokes shift of different benzene-like solutes. The effect of the size of change in partial charges of the solute, the multipolar order of charge distribution, the direction of change, as well as the influence of different solvents on the validity of linear response theory is examined by simulating 54 different systems. Calculation of the Gaussian character of the energy distribution in equilibrium, as well as the time-evolution of the peak width in the nonequilibrium simulation sheds light on the validity of Gaussian statistics in a nonstationary regime. We observed that a large intermediate broadening of the width of the energy distribution correlates with a failure of correlation functions to describe the nonequilibrium event. These results are accompanied by analysis of higher order correlation functions, as well as the structure of the solvents water, acetonitrile and methanol around the solute, to yield a comprehensive view, as well as general guidelines, on when and why equilibrium solvent fluctuations can correctly depict solvation dynamics.
 Please wait while we load your content...
Please wait while we load your content...

