
Effect of protonation on the solvation structure of solute N-butylamine in an aprotic ionic liquid†
 ‡*b
‡*b
Abstract
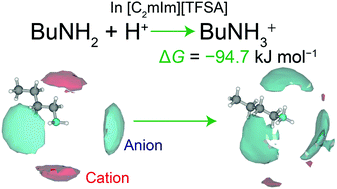
We report on the acid–base reaction of an amine solute in an aprotic ionic liquid from a structural point of view. Thus, the solvation structures of n-butylamine and n-butylammonium (BuNH2 and BuNH3+, respectively, acid–base reaction: BuNH2 + H+ ⇄ BuNH3+) in 1-methyl-3-ethylimidazolium bis(trifluoromethanesulfonyl)amide ([C2mIm][TFSA]) were investigated by high-energy X-ray total scattering combined with molecular dynamics simulations. We found that the solvation structure drastically changed as a result of the protonation reaction in [C2mIm][TFSA]. The NH3+ group was preferentially solvated by TFSA− anions in the protonated BuNH3+ system, whereas the neutral NH2 group was surrounded by both TFSA− and C2mIm+ ions in the BuNH2 system. With regard to the nearest neighbor solute–TFSA interaction, the solvation number for TFSA− anions (nTFSA) increased upon protonation (i.e. BuNH2:nTFSA = 2 and BuNH3+:nTFSA = 3). Both BuNH2 and BuNH3+ interacted with O atoms within TFSA− through hydrogen bonding (i.e. N–H⋯O) interactions, and the N–H⋯O distance was appreciably shorter for positively charged BuNH3+ as compared to neutral BuNH2. These results revealed that the solvation is more stable in energy for the protonated BuNH3+ group because of its stronger hydrogen bonding as compared to neutral BuNH2. This is the origin of the large Gibbs energy for the protonation reaction (ΔG = −94.7 kJ mol−1) and the high acid dissociation constant (pKa = 16.6) of BuNH2 in [C2mIm][TFSA] solution.
 Please wait while we load your content...
Please wait while we load your content...

