
A stochastic method for asphaltene structure formulation from experimental data: avoidance of implausible structures†

Abstract
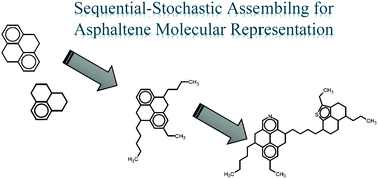
This work presents a stochastic procedure designed to formulate a discrete set of molecular structures that, as a whole, adjust properly to experimental asphaltene data. This algorithm incorporates the pentane effect concept and Clar's sextet rule to the formulation process. The set of viable structures was constructed based on probability distribution functions obtained from experimental information and an isomer database containing all plausible configurations for a given number of rings, avoiding high-energy structures. This procedure was applied to a collection of experimental data from the literature. Ten sets, consisting of 5000 structures each, were obtained. Each set was then optimized. For the most accurate representation, four molecules were sufficient to properly reproduce the experimental input. The asphaltene system obtained is consistent with the reported molecular weight, number of aromatic rings and heteroatom content. Molecular dynamic simulations showed that the asphaltene representation adequately reproduced asphaltene aggregation behavior in toluene and n-heptane. In toluene, a single three-molecule aggregate was observed, and the majority of asphaltene molecules remained in a monomeric state. In n-heptane, aggregates containing up to four molecules were observed; both porous and compact aggregates were found. The asphaltene molecular representation obtained, which allows researchers to avoid inappropriate torsions in the molecule, is able to reproduce interplanar distances between aromatic cores of 4 Å or less for the aggregation state, as supported by experimental results.
 Please wait while we load your content...
Please wait while we load your content...

