
Theoretical study of the charge transport mechanism in π-stacked systems of organic semiconductor crystals†
 *a
*a
Abstract
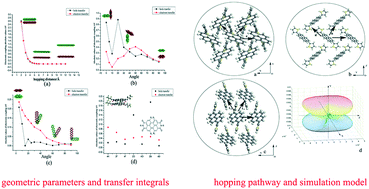
In this study, the orientational dependence of the charge transport mechanism and some important factors related to crystal structure are systematically investigated by quantum chemical methods. Organic semiconductors with small reorganization energies and large transfer integrals originating from their π-stacking crystal structures show excellent charge transport properties. The reorganization energy from the geometrical relaxation occurs during the charge transfer process. The transfer integral of a molecular crystal should be attributed to multiple stacking parameters. A semi-classical simulation model to calculate the anisotropic mobility of a crystalline molecule is extended from one to three dimensions. As we predicted, the anisotropic mobility calculated from our model is improved by considering the contribution from every direction in space, rather than just one plane. This theoretical study determines the importance of tuning the molecular geometry and calculation accuracy for high-performance organic semiconductor materials.
 Please wait while we load your content...
Please wait while we load your content...

