
Using experimental and computational energy equilibration to understand hierarchical self-assembly of Fmoc-dipeptide amphiphiles†

Abstract
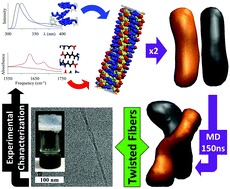
Despite progress, a fundamental understanding of the relationships between the molecular structure and self-assembly configuration of Fmoc-dipeptides is still in its infancy. In this work, we provide a combined experimental and computational approach that makes use of free energy equilibration of a number of related Fmoc-dipeptides to arrive at an atomistic model of Fmoc-threonine-phenylalanine-amide (Fmoc-TF-NH2) which forms twisted fibres. By using dynamic peptide libraries where closely related dipeptide sequences are dynamically exchanged to eventually favour the formation of the thermodynamically most stable configuration, the relative importance of C-terminus modifications (amide versus methyl ester) and contributions of aliphatic versus aromatic amino acids (phenylalanine F vs. leucine L) is determined (F > L and NH2 > OMe). The approach enables a comparative interpretation of spectroscopic data, which can then be used to aid the construction of the atomistic model of the most stable structure (Fmoc-TF-NH2). The comparison of the relative stabilities of the models using molecular dynamic simulations and the correlation with experimental data using dynamic peptide libraries and a range of spectroscopy methods (FTIR, CD, fluorescence) allow for the determination of the nanostructure with atomistic resolution. The final model obtained through this process is able to reproduce the experimentally observed formation of intertwining fibres for Fmoc-TF-NH2, providing information of the interactions involved in the hierarchical supramolecular self-assembly. The developed methodology and approach should be of general use for the characterization of supramolecular structures.
 Please wait while we load your content...
Please wait while we load your content...

