
The IPEA dilemma in CASPT2†

Abstract
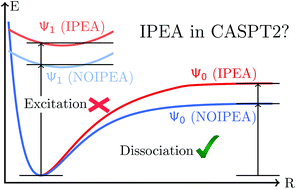
Multi-configurational second order perturbation theory (CASPT2) has become a very popular method for describing excited-state properties since its development in 1990. To account for systematic errors found in the calculation of dissociation energies, an empirical correction applied to the zeroth-order Hamiltonian, called the IPEA shift, was introduced in 2004. The errors were attributed to an unbalanced description of open-shell versus closed-shell electronic states and is believed to also lead to an underestimation of excitation energies. Here we show that the use of the IPEA shift is not justified and the IPEA should not be used to calculate excited states, at least for organic chromophores. This conclusion is the result of three extensive analyses. Firstly, we survey the literature for excitation energies of organic molecules that have been calculated with the unmodified CASPT2 method. We find that the excitation energies of 356 reference values are negligibly underestimated by 0.02 eV. This value is an order of magnitude smaller than the expected error based on the calculation of dissociation energies. Secondly, we perform benchmark full configuration interaction calculations on 137 states of 13 di- and triatomic molecules and compare the results with CASPT2. Also in this case, the excited states are underestimated by only 0.05 eV. Finally, we perform CASPT2 calculations with different IPEA shift values on 309 excited states of 28 organic small and medium-sized organic chromophores. We demonstrate that the size of the IPEA correction scales with the amount of dynamical correlation energy (and thus with the size of the system), and gets immoderate already for the molecules considered here, leading to an overestimation of the excitation energies. It is also found that the IPEA correction strongly depends on the size of the basis set. The dependency on both the size of the system and of the basis set, contradicts the idea of a universal IPEA shift which is able to compensate for systematic CASPT2 errors in the calculation of excited states.
 Please wait while we load your content...
Please wait while we load your content...

