
Substrate selectivity of an isolated enoyl reductase catalytic domain from an iterative highly reducing fungal polyketide synthase reveals key components of programming†
 *ab
*ab
Abstract
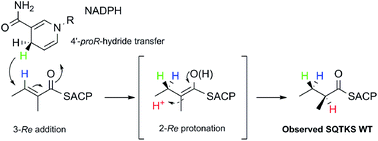
A cis-acting enoyl reductase (ER) catalytic domain was isolated from a fungal highly reducing iterative polyketide synthase (HR-iPKS) for the first time and studied in vitro. The ER from the squalestatin tetraketide synthase forms a discrete dimeric protein in solution. The ER shows broad substrate selectivity, reducing enoyl species including both natural and unnatural substrates. Pantetheine-bound substrate thiolesters reacted much faster than the corresponding SNAC thiolesters. The unnatural substrates included Z-olefins, 2-ethyl olefins and pentaketides. Methylation of the substrate modifies the activity of the ER such that the 2,4-dimethyl oct-2-enoyl substrate fits into the active site but cannot be reduced. A new NMR-based assay was developed for the direct observation of the stereochemical preferences at the 4′ position of the NADPH cofactor and the C-2 and C-3 positions of the substrates. The assay reveals that the fungal iPKS ER-catalysed reaction is stereochemically identical to that of the vertebrate FAS (vFAS) at the cofactor 4′ position and the substrate 3-position, but the high stereoselectivity displayed by intact SQTKS is lost such that reprotonation at the 2-position is unselective by the isolated ER. A 3D model of ER was consistent with these observations and showed that the ER may sequester its final substrate to prevent further chain extension. The results support a developing model for programming by HR-iPKS in which competition for substrates between restrictive and permissive catalytic domains chaperones the growing polyketide to completion, while allowing for errors and evolution.
 Please wait while we load your content...
Please wait while we load your content...

