
The stereodivergent formation of 2,6-cis and 2,6-trans-tetrahydropyrans: experimental and computational investigation of the mechanism of a thioester oxy-Michael cyclization†

Abstract
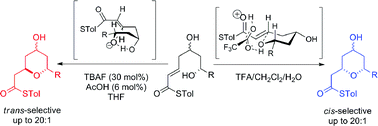
The origins of the stereodivergence in the thioester oxy-Michael cyclization for the formation of 4-hydroxy-2,6-cis- or 2,6-trans-substituted tetrahydropyran rings under different conditions was investigated both computationally and experimentally. Synthetic studies showed that the 4-hydroxyl group was essential for stereodivergence. When the 4-hydroxyl group was present, TBAF-mediated conditions gave the 2,6-trans-tetrahydropyran and trifluoroacetic acid-mediated conditions gave the 2,6-cis-tetrahydropyran. This stereodivergence vanished when the hydroxyl group was removed or protected. Computational studies revealed that: (i) the trifluoroacetic acid catalysed formation of 2,6-cis-tetrahydropyrans was mediated by a trifluoroacetate-hydroxonium bridge and proceeded via a chair-like transition state; (ii) the TBAF-mediated formation of 2,6-trans-tetrahydropyrans proceeded via a boat-like transition state, where the 4-hydroxyl group formed a crucial hydrogen bond to the cyclizing alkoxide; (iii) both reactions are under kinetic control. The utility of this stereodivergent approach for the formation of 4-hydroxy-2,6-substituted tetrahydropyran rings has been demonstrated by the total syntheses of the anti-osteoporotic natural products diospongin A and B.
 Please wait while we load your content...
Please wait while we load your content...

