
Thermal stability of phenolic resin: new insights based on bond dissociation energy and reactivity of functional groups†
Abstract
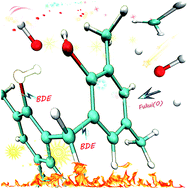
Density functional theory (DFT) was applied to model molecules of phenolic resin (PR) to interpret the relationship between the atomistic structure and thermal properties of the cured PR. The bond dissociation energy (BDE) of C–C (in methylene bridges) and C–O bonds (in hydroxyls), as well as the Fukui function of hydroxyl, benzene and methylene groups, were calculated. The isomers of bisphenol-F and the methyl substituents have a slight effect on the BDEs of C–C and C–O bonds, while the oxidized structures, such as p-benzoquinone and aldehyde groups, lead to a drastic decrease in the C–C bond BDEs. The high reactivity of the carbon atoms in the benzene groups and the oxidized structures results in an increased possibility of being attacked by free radicals and protects the methylenes from being attacked, but it will also lead to ring-opening reactions and weight loss. These results provide a great opportunity to understand the relationship between the atomistic structure and the thermal stability of the cured PR, which plays a pivotal role in the design and optimization of thermal stable polymers.
 Please wait while we load your content...
Please wait while we load your content...

