
Differences between carbon suboxide and its heavier congeners as ligands in transition metal complexes: a theoretical study†
Abstract
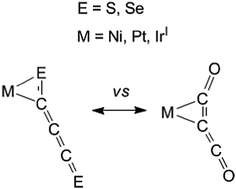
A density functional theory study of experimentally reported [M](C3E2) complexes ([M] = Ir, Ni, Pt surrounded by ligands such as phosphines and halides; E = O, S, Se) suggests that the behaviors of C3O2 and C3E2 (E = S, Se) as ligands in transition metal complexes differ significantly. The lowest energy structures for the [M](C3O2) complexes have the C3O2 ligand bonded to the transition metal through a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. However, the lowest energy structures for the [M](C3E2) (E = S, Se) complexes have the C3E2 ligand bonded to the transition metal through a CE double bond. The latter structures are confirmed by comparison of the experimental infrared frequencies arising from the C3E2 ligand with our theoretical results.
C double bond. However, the lowest energy structures for the [M](C3E2) (E = S, Se) complexes have the C3E2 ligand bonded to the transition metal through a CE double bond. The latter structures are confirmed by comparison of the experimental infrared frequencies arising from the C3E2 ligand with our theoretical results.
 Please wait while we load your content...
Please wait while we load your content...

