
Tumor cell resistance against targeted therapeutics: the density of cultured glioma tumor cells enhances Stat3 activity and offers protection against the tyrosine kinase inhibitor canertinib†
Abstract
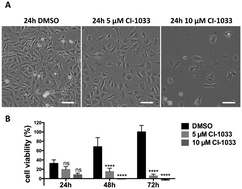
Tumor cell resistance to drug treatment severely limits the therapeutic success of treatment. Tumor cells, exposed to chemotherapeutic drugs, have developed intricate strategies to escape the cytotoxic effects and adapt to adverse conditions. The molecular mechanisms causing drug resistance can be based upon modifications of drug transport or metabolism, structural alterations of drug targets or adaptation of cellular signaling. An important component in the transformation of cells and the emergence of drug resistance is the activation of the transcription factor Stat3. The persistent, inappropriate activation of Stat3 causes the expression of target genes which promote tumor cell proliferation, survival, invasion and immune suppression, and it is instrumental in the process of the emergence of resistance to both conventional chemotherapeutic agents and novel targeted compounds. For these reasons, Stat3 inhibition is being pursued as a promising therapeutic strategy. We have investigated the effects of the tyrosine kinase inhibitor canertinib on the glioma cell line Tu-2449. In these cells Stat3 is persistently phosphorylated and activated downstream of the oncogenic driver v-Src and its effector, the cytoplasmic tyrosine kinase Bmx. Canertinib exposure of Tu-2449 cells rapidly caused the inhibition of the Bmx kinase and the deactivation of Stat3. Prolonged exposure of the cells to canertinib caused the death of the large majority of the cells. Only a few cells became resistant to canertinib and survived in tight clusters. These cells have become drug resistant. When the canertinib resistant cells were expanded and cultured at lower cell densities, they regained their sensitivity towards canertinib. We measured the extent of Stat3 activation as a function of cell density and found that higher cell densities are accompanied by increased Stat3 activation and a higher expression of Stat3 target genes. We suggest that Stat3 induction through tight cell–cell interactions, most likely through the engagement of cadherins, can counteract the inhibitory effects exerted by canertinib on Bmx. Cell–cell interactions induced Stat3 and compensated for the suppression of Stat3 by canertinib, thus transiently protecting the cells from the cytotoxic effects of the inhibitor.
 Please wait while we load your content...
Please wait while we load your content...