
Robust and accurate prediction of protein self-interactions from amino acids sequence using evolutionary information
Abstract
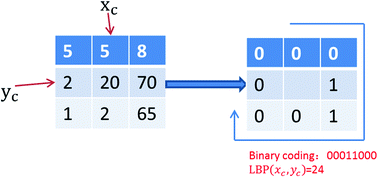
Self-interacting proteins (SIPs) play an essential role in cellular functions and the evolution of protein interaction networks (PINs). Due to the limitations of experimental self-interaction proteins detection technology, it is a very important task to develop a robust and accurate computational approach for SIPs prediction. In this study, we propose a novel computational method for predicting SIPs from protein amino acids sequence. Firstly, a novel feature representation scheme based on Local Binary Pattern (LBP) is developed, in which the evolutionary information, in the form of multiple sequence alignments, is taken into account. Then, by employing the Relevance Vector Machine (RVM) classifier, the performance of our proposed method is evaluated on yeast and human datasets using a five-fold cross-validation test. The experimental results show that the proposed method can achieve high accuracies of 94.82% and 97.28% on yeast and human datasets, respectively. For further assessing the performance of our method, we compared it with the state-of-the-art Support Vector Machine (SVM) classifier, and other existing methods, on the same datasets. Comparison results demonstrate that the proposed method is very promising and could provide a cost-effective alternative for predicting SIPs. In addition, to facilitate extensive studies for future proteomics research, a web server is freely available for academic use at http://219.219.62.123:8888/HASIPP.
 Please wait while we load your content...
Please wait while we load your content...