
Sticking to (first) principles: quantum molecular dynamics and Bayesian probabilistic methods to simulate aquatic pollutant absorption spectra†
Abstract
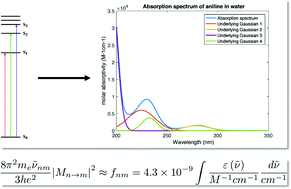
This work explores the relationship between theoretically predicted excitation energies and experimental molar absorption spectra as they pertain to environmental aquatic photochemistry. An overview of pertinent Quantum Chemical descriptions of sunlight-driven electronic transitions in organic pollutants is presented. Second, a combined molecular dynamics (MD), time-dependent density functional theory (TD-DFT) analysis of the ultraviolet to visible (UV-Vis) absorption spectra of six model organic compounds is presented alongside accurate experimental data. The functional relationship between the experimentally observed molar absorption spectrum and the discrete quantum transitions is examined. A rigorous comparison of the accuracy of the theoretical transition energies (ΔES0→Sn) and oscillator strength (fS0→Sn) is afforded by the probabilistic convolution and deconvolution procedure described. This method of deconvolution of experimental spectra using a Gaussian Mixture Model combined with Bayesian Information Criteria (BIC) to determine the mean (μ) and standard deviation (σ) as well as the number of observed singlet to singlet transition energy state distributions. This procedure allows a direct comparison of the one-electron (quantum) transitions that are the result of quantum chemical calculations and the ensemble of non-adiabatic quantum states that produce the macroscopic effect of a molar absorption spectrum. Poor agreement between the vertical excitation energies produced from TD-DFT calculations with five different functionals (CAM-B3LYP, PBE0, M06-2X, BP86, and LC-BLYP) suggest a failure of the theory to capture the low energy, environmentally important, electronic transitions in our model organic pollutants. However, the method of explicit-solvation of the organic solute using the quantum Effective Fragment Potential (EFP) in a density functional molecular dynamics trajectory simulation shows promise as a robust model of the hydrated organic pollutant. Furthermore, the described protocol can be extended using higher-level equilibration and vertical excitation methods to increase the numerical accuracy and describe multi-reference electronic transitions. Finally, a measure of the accuracy of theoretically derived absorption spectra is discussed as a tool to further develop our capacity to produce accurate a priori simulations of sunlight-driven photochemistry in natural waters.
- This article is part of the themed collections: Editor’s Choice: Underappreciated Science and Emerging Investigators 2016
 Please wait while we load your content...
Please wait while we load your content...

