
CO2 adsorption-induced structural changes in coordination polymer ligands elucidated via molecular simulations and experiments†

Abstract
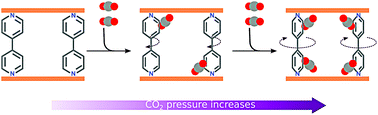
Aiming to elucidate guest-induced structural changes in the coordination polymer CPL-2, grand canonical Monte Carlo (GCMC) simulations were used to predict CO2 loadings in this material, and the results were compared with experimental isotherms. Our calculations suggest that CPL-2 exhibits more pronounced CO2-induced structural changes than previously reported. As the initial evidence, the isotherm simulated in the previously reported CPL-2 structure (experimentally resolved from X-ray diffraction in the “as-synthesized” CPL-2) underestimated the measured CO2 loadings at high pressure, indicating that CPL-2 might undergo structural changes that enable higher pore volumes at high pressure. GCMC simulations in CPL-2 structures considering moderate unit cell expansions reported in the literature still underestimated high-pressure experimental loadings. However, considering an incremental rotation of the CPL-2 bipyridyl pillars with increasing CO2 pressure, we were able to trace the measured isotherm with the simulation data. Computational analysis shows that ligand rotation in CPL-2 enables higher pore volumes, which, in turn, accommodate more CO2 as the gas pressure increases. Desorption measurements suggest that hysteresis in the CO2 isotherm of CPL-2 may also be linked to ligand rotation, and the measured adsorption/desorption cycles show that the rotation is reversible. Based on our simulations for CPL-4 and CPL-5 and previously reported experimental data, it is likely that these materials, which differ from CPL-2 in the bipyridyl ligand, behave similarly in the presence of CO2. Our results help understand the behavior of these materials, which present the kind of structural changes that could be potentially exploited to enhance the CO2 working capacity of ultra-microporous materials for carbon capture applications.
 Please wait while we load your content...
Please wait while we load your content...

