
Dehalogenation of chloroalkanes by nickel(i) porphyrin derivatives, a computational study†
Abstract
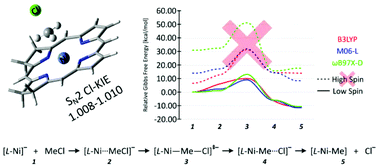
The nickel(I) octaethylisobacteriochlorin anion ([OEiBCh-Ni(I)]−) is commonly used as a synthetic model of cofactor F430 from Methyl-Coenzyme M Reductase. In this regard, experimental studies show that [OEiBCh-Ni(I)]− can catalyze dehalogenation of aliphatic halides in DMF solution by a highly efficient SN2 reaction. To better understand this process, we constructed theoretical models of the dehalogenation of chloromethane by a simple nickel(I) isobacteriochlorin anion and compared its reactivity with that of similar Ni(I) complexes with other porphyrin-derived ligands: porphyrin, chlorin, bactreriochlorin, hexahydroporphyrin and octahydroporphyrin. Our calculations predict that all of the porphyrin derivative's model reactions proceed through low-spin complexes. Relative to the energy of the separate reactants the theoretical activation energies (free-energy barriers with solvation corrections) for the dehalogenation of chloromethane are similar for all of the porphyrin derivatives and range for the different functionals from 10–15 kcal mol−1 for B3LYP to 5–10 kcal mol−1 for M06-L and to 13–18 kcal mol−1 for ωB97X-D. The relative free energies of the products of the dehalogenation step, L-Ni–Me adducts, have a range from −5 to −40 kcal mol−1 for all functionals; generally becoming more negative with increasing saturation of the porphyrin ligand. Moreover, no significant differences in the theoretical chlorine kinetic isotope effect were discernable with change of porphyrin ligand.
 Please wait while we load your content...
Please wait while we load your content...

