
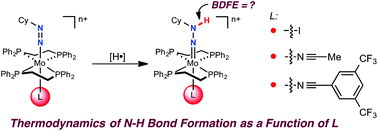
Thermodynamics of N–H bond formation in bis(phosphine) molybdenum(ii) diazenides and the influence of the trans ligand†
Abstract
A series of bis(phosphine) molybdenum(II) diazenides [(dppe)2Mo(NNCy)(I)], [(dppe)2(CH3CN)Mo(NNCy)][BArF24] and [(dppe)2)(3,5-(CF3)2C6H3CN)Mo(NNCy)][BArF24] (dppe = 1,2-bis(diphenylphosphino)ethane; Cy = cyclohexyl; ArF24 = (3,5-(CF3)2C6H3)4) were synthesized and structurally characterized. Treatment of the diazenido complexes with a stoichiometric amount of [H(OEt2)2][BArF24] afforded the corresponding molybdenum(IV) hydrazido species [(dppe)2Mo(NNHCy)(I)][BArF24], [(dppe)2(CH3CN)Mo(NNHCy)][BArF24]2 and [(dppe)2(3,5-(CF3)2C6H3CN)Mo(NNHCy)][BArF24]2, enabling the study of N–H bond dissociation free energies (BDFEs) in the classical Chatt-type bis(phosphine) diazenide platform as a function of ligand (L) trans to the nitrogenous fragment. Deprotonation and electrochemical experiments established that the trans nitrile 3,5-(CF3)2C6H3CN afforded the least reducing molybdenum(IV) hydrazido complex in the series (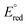 = −1.32 V vs. Fc/Fc+) with the most acidic N–H bond (pKa < 2.6, THF), whereas the ligands CH3CN (
= −1.32 V vs. Fc/Fc+) with the most acidic N–H bond (pKa < 2.6, THF), whereas the ligands CH3CN (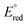 = −1.60 V, pKa < 5.5) and I− (
= −1.60 V, pKa < 5.5) and I− (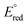 = −2.03 V, pKa = 9.3) gave more reducing complexes with less acidic N–H bonds. Computational (DFT) studies confirm weak N–H bond strengths of 32.8 (L = I−), 35.4 (L = CH3CN) and 36.2 kcal mol−1 (L = 3,5-(CF3)2C6H3CN) in the hydrazido series.
= −2.03 V, pKa = 9.3) gave more reducing complexes with less acidic N–H bonds. Computational (DFT) studies confirm weak N–H bond strengths of 32.8 (L = I−), 35.4 (L = CH3CN) and 36.2 kcal mol−1 (L = 3,5-(CF3)2C6H3CN) in the hydrazido series.
- This article is part of the themed collections: Recipients of the Dalton Transactions UC Berkeley Lecture and Reactions Facilitated by Ligand Design
 Please wait while we load your content...
Please wait while we load your content...

