
Unimolecular decomposition pathways of negatively charged nitriles by ab initio molecular dynamics
Abstract
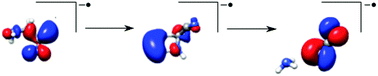
We present negative ion-mode simulations within the QCEIMS program [Grimme, Angew. Chem., Int. Ed., 2013, 52, 6306]. It is an exhaustive and robust ab initio molecular dynamics/stochastic algorithm used to perform simulations of unimolecular decomposition of anions, in unprecedented detail. The objective of this approach is to compliment electron attachment spectroscopy and aid in the interpretation of relevant dissociation dynamics. Prototypical simulations are performed for the four nitrile compounds acetonitrile, cyanamide, aminoacetonitrile, and trifluoroacetonitrile. The unique decomposition pathways which naturally occur in the simulations are addressed along with fractional yields, reaction times and relative intensities of the fragments. Furthermore, trajectories of selected decomposition pathways of the aminoacetonitrile anion are investigated in greater detail, where we find that the relevant HOMO of the anion has a mixed π* and σ* character delocalized over the entire molecule.
 Please wait while we load your content...
Please wait while we load your content...

