
Double quantum coherence ESR spectroscopy and quantum chemical calculations on a BDPA biradical†
Abstract
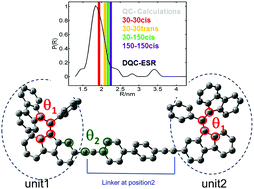
Carbon-centered radicals are interesting alternatives to otherwise commonly used nitroxide spin labels for dipolar spectroscopy techniques because of their narrow ESR linewidth. Herein, we present a novel BDPA biradical, where two BDPA (α,α,γ,γ-bisdiphenylene-β-phenylallyl) radicals are covalently tethered by a saturated biphenyl acetylene linker. The inter-spin distance between the two spin carrier fragments was measured using double quantum coherence (DQC) ESR methodology. The DQC experiment revealed a mean distance of only 1.8 nm between the two unpaired electron spins. This distance is shorter than the predictions based on a simple modelling of the biradical geometry with the electron spins located at the central carbon atoms. Therefore, DFT (density functional theory) calculations were performed to obtain a picture of the spin delocalization, which may give rise to a modified dipolar interaction tensor, and to find those conformations that correspond best to the experimentally observed inter-spin distance. Quantum chemical calculations showed that the attachment of the biphenyl acetylene linker at the second position of the fluorenyl ring of BDPA did not affect the spin population or geometry of the BDPA radical. Therefore, spin delocalization and geometry optimization of each BDPA moiety could be performed on the monomeric unit alone. The allylic dihedral angle θ1 between the fluorenyl rings in the monomer subunit was determined to be 30° or 150° using quantum chemical calculations. The proton hyperfine coupling constant calculated from both energy minima was in very good agreement with literature values. Based on the optimal monomer geometries and spin density distributions, the dipolar coupling interaction between both BDPA units could be calculated for several dimer geometries. It was shown that the rotation of the BDPA units around the linker axis (θ2) does not significantly influence the dipolar coupling strength when compared to the allylic dihedral angle θ1. A good agreement between the experimental and calculated dipolar coupling was found for θ1 = 30°.
 Please wait while we load your content...
Please wait while we load your content...

