
Enhanced conformational sampling technique provides an energy landscape view of large-scale protein conformational transitions†
Abstract
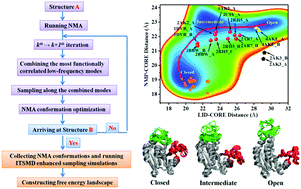
Large-scale conformational changes in proteins are important for their functions. Tracking the conformational change in real time at the level of a single protein molecule, however, remains a great challenge. In this article, we present a novel in silico approach with the combination of normal mode analysis and integrated-tempering-sampling molecular simulation (NMA–ITS) to give quantitative data for exploring the conformational transition pathway in multi-dimensional energy landscapes starting only from the knowledge of the two endpoint structures of the protein. The open-to-closed transitions of three proteins, including nCaM, AdK, and HIV-1 PR, were investigated using NMA–ITS simulations. The three proteins have varied structural flexibilities and domain communications in their respective conformational changes. The transition state structure in the conformational change of nCaM and the associated free-energy barrier are in agreement with those measured in a standard explicit-solvent REMD simulation. The experimentally measured transition intermediate structures of the intrinsically flexible AdK are captured by the conformational transition pathway measured here. The dominant transition pathways between the closed and fully open states of HIV-1 PR are very similar to those observed in recent REMD simulations. Finally, the evaluated relaxation times of the conformational transitions of three proteins are roughly at the same level as reported experimental data. Therefore, the NMA–ITS method is applicable for a variety of cases, providing both qualitative and quantitative insights into the conformational changes associated with the real functions of proteins.
 Please wait while we load your content...
Please wait while we load your content...

