
Adsorption of water and ethanol on noble and transition-metal substrates: a density functional investigation within van der Waals corrections†
Abstract
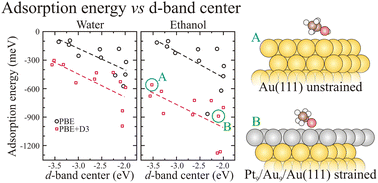
We report the results of extensive computational investigation of the adsorption properties of water and ethanol on several Cu-, Pt-, and Au-based substrates, including the close-packed unreconstructed Cu(111), Pt(111), and Au(111) surfaces, defected metal substrates with on-surface low-coordinated sites generated by the intermixing of Pt–Cu and Pt–Au in the topmost surface layers and strained on-surface and sub-surface Pt-layers at Cu(111) and Au(111) substrates. The calculations are based on the density functional theory (DFT) within the van der Waals (vdW) correction. For all the substrates, we found that water and ethanol bind via the anionic O atom to the cationic one-fold coordinated on-top metal sites, which enhances the adsorbate–substrate Coulomb interactions. For water, both DFT and DFT + vdW calculations predict a flat geometry. For ethanol, the DFT and DFT + vdW results are in contrast, namely, DFT yields a perpendicular orientation of the C–C bond with respect to the surface, while we obtained a parallel orientation of the C–C bond using DFT + vdW, which maximizes the adsorption energies. Despite expected deviations due to the nature of the weak adsorbate–substrate interactions, we found that the adsorption energy of water and ethanol shows a linear dependence as a function of the position of the center of gravity of the occupied d-band, and hence, the magnitude of the adsorption energy increases as the d-band center position shifts towards the Fermi energy. Thus, it indicates hybridization between the O p- and metal d-states, which determines the magnitude of the adsorption energy of water and ethanol on clean, low-coordinated, and strained noble and transition-metal substrates.
 Please wait while we load your content...
Please wait while we load your content...

