
A DFT kinetic study on 1,3-dipolar cycloaddition reactions in solution†
Abstract
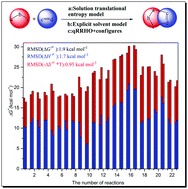
Several popular density functional theory (DFT) methods have been employed to characterize a series of 1,3-dipolar cycloaddition reactions, including the exploration of reaction mechanisms and the calculations of kinetic parameters. Both the gas- and solution-phase translational entropy models have been used to calculate the activation entropies, and the results obtained from the latter method are quite close to the experimental measurements. For some of the reactions studied, e.g., a1 + b9, a1 + b10, a5 + b9 and a12 + b5, the explicit + implicit solvation model, namely, a cluster-continuum type model, should be employed to account for the specific solvent–solute interactions. The quasi rigid-rotor-harmonic-oscillator (qRRHO) small frequency vibrational entropy correction, in conjunction with the conformational entropy correction, could further improve the calculated activation entropy data. The comparison between calculation data with experimental measurements, using 23 activation entropies and 160 reaction rate constants as test benchmark, demonstrated that our present strategy could calibrate the root-mean-square-deviation (RMSD) of activation entropies to be 1.8 cal mol−1 K−1 and that of Gibbs free energy barriers to be 1.8 kcal mol−1.
 Please wait while we load your content...
Please wait while we load your content...

