
Conformational features of the Aβ42 peptide monomer and its interaction with the surrounding solvent†
Abstract
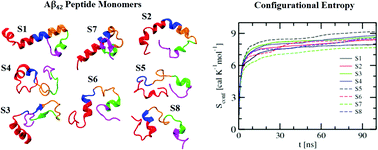
Accumulation of the amyloid beta (Aβ) peptide in the brain is responsible for debilitating neurodegenerative diseases, such as Alzheimer's disease (AD). We have carried out atomistic molecular dynamics simulations of the full-length Aβ42 peptide monomer with a wide range of conformations at room temperature. Efforts have been made to probe the conformational features of different segments of the peptide, namely the two terminal segments (N-term and C-term), the central hydrophobic regions (hp1 and hp2) and the central turn region joining hp1 and hp2, and their nonuniform influence on the spatial arrangements and binding energies of the surrounding water molecules. Our calculations reveal fluctuating conformations of the monomers with the formation and breaking of different secondary structural elements. In particular, it is noticed that the Aβ monomers exhibit a propensity to either retain or transform into a helical form toward the N-term region and a β-strand-like form near the C-term segment. Besides, heterogeneous conformational flexibility of the Aβ monomers has been found to be correlated with the corresponding nonuniform entropy gains. Additionally, our calculation further reveals a heterogeneous hydration environment around the peptide. It is found that irrespective of the Aβ peptide conformations and their nonuniform fluctuations, water molecules around the hydrophobic hp1 and hp2 segments are relatively weakly bound. This is an important observation, as in the presence of other monomers such weakly bound water molecules around hp1 and hp2 are expected to be easily displaced during the hydrophobic collapse that leads to Aβ aggregation.
 Please wait while we load your content...
Please wait while we load your content...

