
Density functional theory simulation of the adsorption of sulphur multilayers on Au(100)†
Abstract
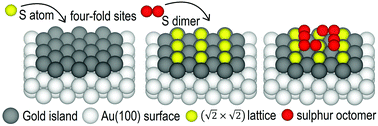
The adsorption of sulphur multilayers on Au(100) has been studied using density functional theory (DFT) within the generalized gradient approximation (GGA). The first sulphur layer was adsorbed on the four-fold sites of the unreconstructed Au(100) surface forming a 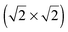 lattice. The experimental parameters of the
lattice. The experimental parameters of the 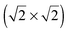 lattice were reproduced taking into account the surface expansion of the topmost Au(100) layer. This expansion should occur when gold islands are formed after the lifting of hex-reconstruction, which allows the lateral movement of the gold atoms. The second sulphur layer, on top of the
lattice were reproduced taking into account the surface expansion of the topmost Au(100) layer. This expansion should occur when gold islands are formed after the lifting of hex-reconstruction, which allows the lateral movement of the gold atoms. The second sulphur layer, on top of the 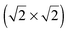 lattice, consisted of eight S atoms (octomer phase) in a quasi-rectangular arrangement. The structural optimization of the octomer phase was achieved in a specific spatial orientation with respect to the
lattice, consisted of eight S atoms (octomer phase) in a quasi-rectangular arrangement. The structural optimization of the octomer phase was achieved in a specific spatial orientation with respect to the 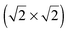 lattice. The analysis of Bader atomic charges and the projected density of states (PDOS) demonstrated that charge transfer from the Au(100) surface to the sulphur layers, sulphur chemisorption and sulphur–sulphur inter-layer mixing of electronic states control the formation of sulphur multilayers.
lattice. The analysis of Bader atomic charges and the projected density of states (PDOS) demonstrated that charge transfer from the Au(100) surface to the sulphur layers, sulphur chemisorption and sulphur–sulphur inter-layer mixing of electronic states control the formation of sulphur multilayers.
 Please wait while we load your content...
Please wait while we load your content...

