
Local NH–π interactions involving aromatic residues of proteins: influence of backbone conformation and ππ* excitation on the π H-bond strength, as revealed from studies of isolated model peptides
Abstract
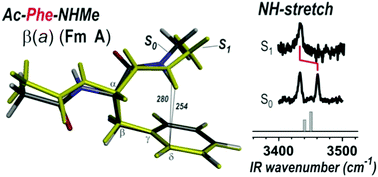
Conformer-selective IR gas phase spectroscopy and high level quantum chemistry methods have been used to characterise the diversity of local NH–π interactions between the π ring of a phenylalanine aromatic residue and the nearby main chain amide groups. The study of model systems shows how the amide NH stretch vibrational features, in the 3410–3460 cm−1 frequency range, can be used to monitor the strength of these local π H-bonds, which is found to depend on both the backbone conformation and the aromatic side chain orientation. This is rationalized in terms of partial electron transfer between the π cloud and the main chain NH bonds, with the help of analysis tools based on Natural Bonding Orbitals and Non-Covalent Interactions plots. The experimental study, extended to the NH–π interactions when the Phe residue is excited in its first ππ* electronic state, also demonstrates the principle of the ππ* labelling technique, i.e. a selective labelling of those NH bonds in a peptide molecule that are in close contact with an aromatic ring, as an elegant tool for IR spectroscopic assignments. The validation of theoretical predictions against experimental data (frequency change upon excitation) eventually qualifies the use of the CC2 method for the description of the ππ* excited states of systems having a phenyl ring, both in terms of structure, vibrational modes and nature of excited states.
 Please wait while we load your content...
Please wait while we load your content...

