
An unusual H2 sorption mechanism in PCN-14: insights from molecular simulation†
Abstract
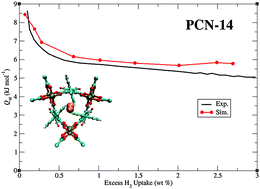
Simulations of H2 sorption were performed in PCN-14, a metal–organic framework (MOF) that consists of Cu2+ ions coordinated to 5,5′-(9,10-anthracenediyl)diisophthalate (adip) linkers. This MOF displays an excess H2 uptake of 2.70 wt% at 77 K and 1.0 atm and an initial H2Qst value of 8.6 kJ mol−1 according to previous experimental measurements. The experimental H2 sorption isotherms and Qst values in PCN-14 were reproduced in simulations using well-known H2 potentials that have been widely used for MOF–H2 theoretical studies. H2 sorption in PCN-14 was dominated by repulsion/dispersion energetics; this allowed the experimental observables to be reproduced by a model that includes only Lennard-Jones parameters. The most energetically favorable H2 sorption site in PCN-14 corresponds to sorption within a small cage that is enclosed by three [Cu2(O2CR)4] units and three adip linkers. The anthracenyl rings of the adip linkers represent the secondary sorption sites within the MOF. In contrast to expectations, sorption of H2 onto the Cu2+ ions of the copper paddlewheels was not observed within the simulations at low loading. The simulations revealed that the open-metal sites in PCN-14 were occupied at high loading. Control simulations of H2 sorption in PCN-14 for different cases in which the partial positive charge of one of the paddlewheel Cu2+ ions was increased relative to the other revealed that sorption onto the open-metal sites can be captured if there is a very high positive charge on the metal. Otherwise, the calculated partial charge for the Cu2+ ions in PCN-14 in this work was not high enough in magnitude to facilitate strong H2–metal interactions in simulation. This study shows the power of using molecular simulations to elucidate an unusual H2 sorption behavior in a MOF.
 Please wait while we load your content...
Please wait while we load your content...

