
The i-TTM model for ab initio-based ion–water interaction potentials. II. Alkali metal ion–water potential energy functions†
Abstract
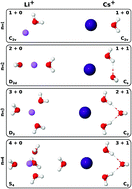
A new set of i-TTM potential energy functions describing the interactions between alkali metal ions and water molecules is reported. Following our previous study on halide ion–water interactions [J. Phys. Chem. B, 2016, 120, 1822], the new i-TTM potentials are derived from fits to CCSD(T) reference energies and, by construction, are compatible with the MB-pol many-body potential, which has been shown to accurately predict the properties of water from the gas to the condensed phase. Within the i-TTM formalism, two-body repulsion, electrostatic, and dispersion energies are treated explicitly, while many-body effects are represented by classical induction. The accuracy of the new i-TTM potentials is assessed through extensive comparisons with results obtained from different ab initio methods, including CCSD(T), CCSD(T)-F12b, DF-MP2, and several DFT models, as well as from polarizable force fields for M+(H2O)n clusters with M+ = Li+, Na+, K+, Rb+, and Cs+, and n = 1–4.
- This article is part of the themed collection: Insights from advanced methods in molecular dynamics
 Please wait while we load your content...
Please wait while we load your content...

