
Elucidation of the conformational dynamics of multi-body systems by construction of Markov state models
Abstract
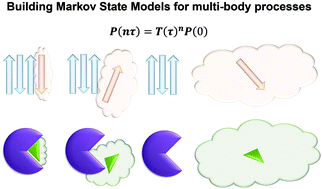
Constructing Markov State Models (MSMs) based on short molecular dynamics simulations is a powerful computational technique to complement experiments in predicting long-time kinetics of biomolecular processes at atomic resolution. Even though the MSM approach has been widely applied to study one-body processes such as protein folding and enzyme conformational changes, the majority of biological processes, e.g. protein–ligand recognition, signal transduction, and protein aggregation, essentially involve multiple entities. Here we review the attempts at constructing MSMs for multi-body systems, point out the challenges therein and discuss recent algorithmic progresses that alleviate these challenges. In particular, we describe an automatic kinetics based partitioning method that achieves optimal definition of the conformational states in a multi-body system, and discuss a novel maximum-likelihood approach that efficiently estimates the slow uphill kinetics utilizing pre-computed equilibrium populations of all states. We expect that these new algorithms and their combinations may boost investigations of important multi-body biological processes via the efficient construction of MSMs.
- This article is part of the themed collections: PCCP Perspectives and Insights from advanced methods in molecular dynamics
 Please wait while we load your content...
Please wait while we load your content...

