
Spectroscopic and computational studies of nitrile hydratase: insights into geometric and electronic structure and the mechanism of amide synthesis†
 *b
and
*b
and
Abstract
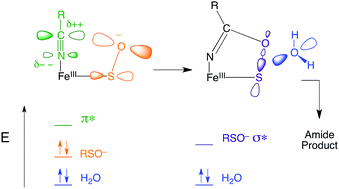
Nitrile hydratases (NHases) are mononuclear nonheme enzymes that catalyze the hydration of nitriles to amides. NHase is unusual in that it utilizes a low-spin (LS) FeIII center and a unique ligand set comprised of two deprotonated backbone amides, cysteine-based sulfenic acid (RSO(H)) and sulfinic acid (RSO2−), and an unmodified cysteine trans to an exogenous ligand site. Electron paramagnetic resonance (EPR), magnetic circular dichroism (MCD) and low-temperature absorption (LT-Abs) spectroscopies are used to determine the geometric and electronic structures of butyrate-bound (NHaseBA) and active (NHaseAq) NHase. These data calibrate DFT models, which are then extended to explore the mechanism of nitrile hydration by NHase. In particular, the nitrile is activated by coordination to the LS FeIII and the sulfenate group is found to be deprotonated and a significantly better nucleophile than water that can attack the coordinated nitrile to form a cyclic species. Attack at the sulfenate S atom of the cyclic species is favorable and leads to a lower kinetic barrier than attack by water on coordinated, uncyclized nitrile, while attack at the C of the cyclic species is unfavorable. The roles of the unique ligand set and low-spin nature of the NHase active site in function are also explored. It is found that the oxidized thiolate ligands are crucial to maintaining the LS state, which is important in the binding and activation of nitrile susbtrates. The dominant role of the backbone amidate ligands appears to be as a chelate in keeping the sulfenate properly oriented for nucleophilic attack on the coordinated substrate.
 Please wait while we load your content...
Please wait while we load your content...

