
Diffusion of alkali metals in the first stage graphite intercalation compounds by vdW-DFT calculations
Abstract
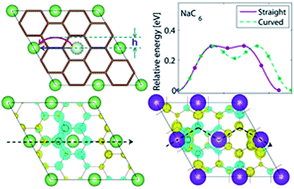
Diffusion of alkali metal cations in the first stage graphite intercalation compounds (GIC) LiC6, NaC6, NaC8 and KC8 has been investigated with density functional theory (DFT) calculations using the optPBE-vdW van der Waals functional. The formation energies of alkali vacancies, interstitials and Frenkel defects were calculated and vacancies were found to be the dominating point defects. The diffusion coefficients of the alkali metals in GIC were evaluated by a hopping model of point defects where the energy barriers for vacancy diffusion were derived from transition state theory. For LiC6, NaC6, NaC8 and KC8, respectively, the diffusion coefficients were found to be 1.5 × 10−15, 2.8 × 10−12, 7.8 × 10−13 and 2.0 × 10−10 m2 s−1 at room temperature, which is within the range of available experimental data. For LiC6 and NaC6 a curved vacancy migration path is the most energetically favourable, while a straight pathway was inferred for NaC8 and KC8. The diffusion coefficients for alkali metal vacancy diffusion in first stage GICs scales with the graphene interlayer spacing: LiC6 ≪ NaC8 < NaC6 ≪ KC8.
 Please wait while we load your content...
Please wait while we load your content...

