
A quantum Monte Carlo study on electron correlation effects in small aluminum hydride clusters†
Abstract
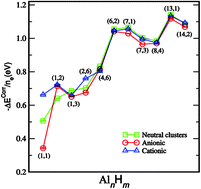
Using the fixed-node diffusion quantum Monte Carlo (DMC) method, we investigate the electron correlation in several small relaxed and unrelaxed neutral, cationic, and anionic aluminum hydride clusters. We calculate the clusters total energies and use them to obtain the binding energies. Our results are in very good agreement with the available ab initio calculations and anion-photoelectron spectroscopy experiments. The calculations have also been performed in the Hartree–Fock (HF) approximation in order to analyse the impact of electron correlation. For the total atomic binding energy, i.e. the energy necessary to separate all the atoms, this impact varies from 20% up to about 50%, whereas for the electron binding energy, i.e. the energy required to detach or attach an electron to the cluster, it ranges from 1% up to 73%. The decomposition of the electron binding energies clearly shows that both charge redistribution and electron correlation are important for determining the detachment energies, whereas electrostatic and exchange interactions are responsible for the ionization potential.
 Please wait while we load your content...
Please wait while we load your content...

