
Distinguishing the influence of structural and energetic disorder on electron transport in fullerene multi-adducts†
Abstract
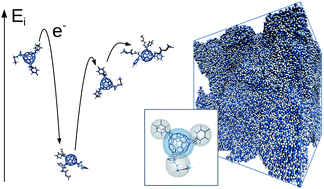
Fullerene multi-adducts offer a method to tune the open-circuit voltage of organic solar cells but generally lead to poor photocurrent generation which has been linked to poor electron transport in the fullerenes. The poor electron transport may result from the effect of multiple side chains in hindering close packing of the fullerene cages or from disorder in energy levels due to the presence of multiple isomers. Here, we present time-of-flight and field-effect transistor measurements of mobility in [6,6]-phenyl-C-61-butyric acid methyl ester (PCBM) and its higher adducts. To understand the origin of the poor electron mobility, we develop a coarse-grained molecular dynamics model to build isomeric mixes of the multi-adducts. The coarse grained model massively speeds up the simulation relative to atomistic molecular dynamics, enabling assemblies of 100 000 molecules to be studied. We simulate electron transport in the structures using a kinetic Monte Carlo method, accounting for variations in site energy using electronic structure calculations. This allows us to separate the influence of packing disorder (poorer contact between fullerenes due to steric hindrance) from the influence of energetic disorder (due to varying acceptor energies of the isomers) on electron mobility. We find that energetic disorder due to different isomers dominates the trend in charge transport. Consequently, pure isomeric samples of higher fullerene adducts should enable higher efficiency solar cells.
 Please wait while we load your content...
Please wait while we load your content...

