
Molecular modelling studies of sirtuin 2 inhibitors using three-dimensional structure–activity relationship analysis and molecular dynamics simulations†
Abstract
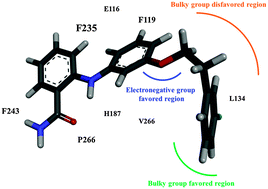
Sirtuin 2 (SIRT2) is a nicotinamide-adenine-dinucleotide-dependent histone deacetylase that plays a vital role in various biological processes related to DNA regulation, metabolism, and longevity. Recent studies on SIRT2 have indicated its therapeutic potential for neurodegenerative diseases such as Parkinson's disease. In this study, a series of SIRT2 inhibitors with a 2-anilinobenzamide core was analysed using a combination of molecular modelling techniques. A three-dimensional structure–activity relationship (3D-QSAR) model adopting a comparative molecular field analysis (CoMFA) method with a non-cross-validated correlation coefficient R2 = 0.992 (for training set) and a correlation coefficient Rtest2 = 0.804 (for test set) was generated to determine the structural requirements for inhibitory activity. Furthermore, we employed molecular dynamics (MD) simulations and the molecular mechanics/generalized Born surface area (MM/GBSA) method to compare the binding modes of a potent and selective compound interacting with SIRT1, SIRT2, and SIRT3 and also their binding free energies to shed light on the selectivity of the footing of structural and energetic investigations. The steric and electrostatic contour maps from the 3D-QSAR analysis identified several key interactions also observed in the MD simulations. According to these results, we provide guidelines for developing novel potent and selective SIRT2 inhibitors.
 Please wait while we load your content...
Please wait while we load your content...