
Pentaborate(1−) salts templated by substituted pyrrolidinium cations: synthesis, structural characterization, and modelling of solid-state H-bond interactions by DFT calculations†

Abstract
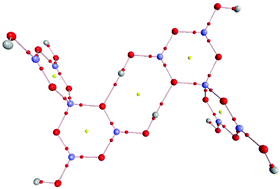
The synthesis and characterization of a series of pentaborate(1−) salts of substituted pyrrolidinium cations [C4H8NH2][B5O6(OH)4] (1), [C4H8NMe2][B5O6(OH)4] (2) [C4H8NMeH][B5O6(OH)4] (3), [(2-CH2OH)C4H7NH2][B5O6(OH)4] (4) is reported. All compounds were characterized by single-crystal XRD studies with 3 (1/2CH3COCH3) and 4 (1/2H2O) solvated. TGA/DSC analysis of the pentaborates 1–4 showed that they thermally decomposed in air at 800 °C to 2.5 B2O3, in a 2 step process involving dehydration (<250 °C) and oxidative decomposition (250–600 °C). BET analysis of materials derived thermally from the pentaborates 1 and 2 had internal porosities of <1 m2 g−1, indicating they were non-porous. All compounds show extensive supramolecular H-bonded anionic lattices. H-bond interactions are described in detail and motifs found in these and in other pentaborate structures have been examined and modelled by DFT calculations. These calculations confirm that H-bonds interactions in pentaborates are moderately strong (ca. −10 to −21 kJ mol−1) and are likely to dominate the energetics of their templated syntheses.
- This article is part of the themed collection: In memory of Professor Kenneth Wade
 Please wait while we load your content...
Please wait while we load your content...

