
Dynamics of ultrathin gold layers on vitreous silica probed by density functional theory
Abstract
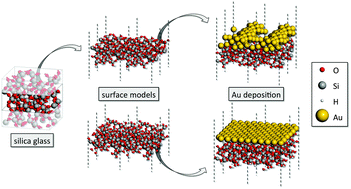
The structure and properties of Au ultrathin films on hydroxyl-free and hydroxylated silica glass surfaces are investigated using ab initio molecular dynamics simulations. Substantial surface structure dependence of Au agglomeration behavior (solid-state dewetting) is found. On hydroxyl-free surfaces, the Au film virtually undergoes instantaneous agglomeration accompanied by the formation of voids exposing a bare silica glass surface. In contrast, simulated annealing of the Au film on hydroxylated surface models leaves its structure unchanged within the simulation time. This points to a key role of reactive defect sites in the kinetics of solid-state dewetting processes of metals deposited on the glass surface. Such sites are important for initial void nucleation and formation of metal clusters. In addition, our calculations demonstrate the crucial role of the appropriate inclusion of dispersion interactions in density functional theory simulations of metals deposited on glass surfaces. For defective, hydroxyl-free glass surfaces the dispersion correction accounts for 35% of the total adhesion energy. The effect is even more dramatic for hydroxylated glass surfaces, where adhesion energies are almost entirely due to dispersion interactions. The Au adhesion energies of 200 and 160 kJ (mol nm2)−1 calculated for hydroxylated glass surfaces are in good agreement with the experimental data.
 Please wait while we load your content...
Please wait while we load your content...

