
DFT-based Green's function pathways model for prediction of bridge-mediated electronic coupling†
Abstract
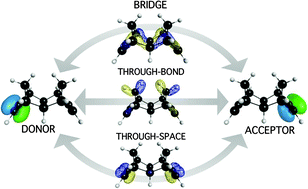
A density functional theory-based Green's function pathway model is developed enabling further advancements towards the long-standing challenge of accurate yet inexpensive prediction of electron transfer rate. Electronic coupling predictions are demonstrated to within 0.1 eV of experiment for organic and biological systems of moderately large size, with modest computational expense. Benchmarking and comparisons are made across density functional type, basis set extent, and orbital localization scheme. The resulting framework is shown to be flexible and to offer quantitative prediction of both electronic coupling and tunneling pathways in covalently bound non-adiabatic donor–bridge–acceptor (D–B–A) systems. A new localized molecular orbital Green's function pathway method (LMO-GFM) adaptation enables intuitive understanding of electron tunneling in terms of through-bond and through-space interactions.
- This article is part of the themed collection: Measurement and prediction of quantum coherence effects in biological processes
 Please wait while we load your content...
Please wait while we load your content...

