
Anomalous doping effect in black phosphorene using first-principles calculations
Abstract
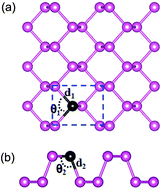
Using first-principles density functional theory calculations, we investigate the geometries, electronic structures, and thermodynamic stabilities of substitutionally doped phosphorene sheets with group III, IV, V, and VI elements. We find that the electronic properties of phosphorene are drastically modified by the number of valence electrons in dopant atoms. The dopants with an even number of valence electrons enable the doped phosphorenes to have a metallic feature, while the dopants with an odd number of valence electrons retain a semiconducting feature. This even–odd oscillating behavior is attributed to the peculiar bonding characteristics of phosphorene and the strong hybridization of sp orbitals between dopants and phosphorene. Furthermore, the calculated formation energies of various substitutional dopants in phosphorene show that such doped systems can be thermodynamically stable. These results propose an intriguing route to tune the transport properties of electronic and photoelectronic devices based on phosphorene.
 Please wait while we load your content...
Please wait while we load your content...

