
Dissociation of oxygen on pristine and nitrogen-doped carbon nanotubes: a spin-polarized density functional study
Abstract
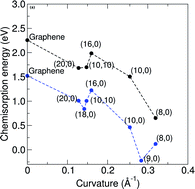
Although nitrogen-doped nanocarbon systems have recently received intense attention, the mechanism for the observed highly efficient oxygen reduction is still under debate. To address this issue, we investigated the adsorption and dissociation of an oxygen molecule on three pristine or nitrogen-doped nanocarbon systems: graphene, single-walled and double-walled carbon nanotubes using density functional theory calculations. The adsorption and dissociation energies were determined for both pristine and N-doped single-walled carbon nanotubes of different diameters with graphitic-like N substitutions in order to see the effect of diameter on oxygen dissociation. It was found that the energy barrier for oxygen dissociation, chemisorption energy and reaction energy are a function of carbon nanotube diameter, but independent of the number of walls. We also investigated the energy barrier of oxygen dissociation on single-walled carbon nanotubes with different types of nitrogen doping (i.e. pyridinic and graphitic). It was observed that higher nitrogen concentrations greatly reduce the energy barrier for graphitic nitrogen. Our results contribute towards a better understanding of the reaction mechanism for nitrogen-doped carbon nanomaterials involving oxygen molecule dissociation in the first step.
 Please wait while we load your content...
Please wait while we load your content...

